BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://journal.zums.ac.ir/article-1-6545-en.html



2- Dept. of Physiology and Pharmacology, Faculty of Medicine, North Khorasan University of Medical Sciences, Bojnourd, Iran
3- Pharmacological Research Center of Medicinal Plants, Mashhad University of Medical Sciences, Mashhad, Iran
4- Pharmacological Research Center of Medicinal Plants, Mashhad University of Medical Sciences, Mashhad, Iran ,
✅ The findings suggest that R. turkestanicum extract has neuroprotective activity on 6-OHDA-induced neuronal toxicity of neuroblastoma cells.
As the most well-known progressive neurodegenerative disorder, Parkinson's disease (PD), is identified by a selected loss of dopaminergic neurons within the brain (1), which results in debilitating symptoms including uncontrollable tremor, bradykinesia, and inflexibility (2). Supplementation with levodopa has shown efficient therapeutic benefits for the treatment of PD. The reason for idiopathic PD is unclear. Prior surveys have shown that various triggers are involved in the pathogenesis of PD, such as generation of free radicals and dysfunction of mitochondrial and natural agents, which lead to toxicity (3).
The amount of oxidative stress is accumulated in the substantia nigra of PD patients (4). The production of MDA following lipid peroxidation and reactive oxygen species [ROS], environmental toxins, and mitochondrial dysfunction are the leading causes of PD (5). Based on these findings, excessive ROS production could be critically related to the pathology of neurodegenerative problems (6, 7). 6-hydroxydopamine (6-OHDA) is a toxin that injures neurons and is classically used in neurodegenerative models as it leads to oxidative damage and cell death of dopaminergic cells. Several studies have indicated that modulation of oxidative stress can decrease the degeneration of neurons following 6-OHDA toxicity, which is utilized to develop neurodegenerative laboratory models (8).
Rheum species is related to the Polygonaceae plant family and is used widely in traditional Chinese medicine. Anthraquinones, phenolic compounds, dianthrones, glycosides, and tannins are active compounds of the Rheum species. R. turkestanicum Janischew grows widely in central Asia and the northeast of Iran (9). The root of R. turkestanicum is a well-known traditional Iranian herbal medicine commonly used to treat hypertension, diabetes, and cancer (10). Different investigations have reported that the species of Rheum, including R. undulatum contains antioxidant compounds (11-13). It is reported that R. undulatum diminishes ROS and hydrogen peroxide (14).
Further studies have indicated that R. emodi reduces the toxicity of cardiac cells against H2O2 (10, 15). Despite the antioxidant nature of R. turkestanicum, no experimental evidence exists to prove the neuroprotective effect of this medicinal herb. Hence, this research evaluated the protective impacts of R. turkestanicum hydroalcoholic extract on 6-OHDA-stimulated neuronal damage against SH-SY5Y. The current outcomes revealed that R. turkestanicum suppressed 6-OHDA-induced neuronal toxicity by inhibiting free radicals and MDA levels in human neuroblastoma in vitro.
Reagents
After receiving approval from the Ethics Committee, the experiments on cells were done according to national guidelines (IR.MUMS.MEDICAL.REC.1397.587). MTT powder (98%) was prepared from Thermo Fisher (Thermo Fisher Scientific, Inc., Waltham, MA, USA). 6-hydroxydopamine (6-OHDA), propidium-iodide (PI), the fluorescent probe 2, 7-dichlorofluorescein diacetate (DCF-DA), dimethyl sulfoxide (DMSO), and phosphate buffer saline (PBS) were acquired from Sigma-Aldrich (St. Louis, MO, USA). RNase A was bought from Alfa Aesar (Ward Hill, MA, USA). High glucose Dulbecco Modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco (Grand Island, NY). Trichloroacetic acid (TCA), TBA (thiobarbituric acid), and DTNB (5,5′-Dithiobis(2-nitrobenzoic acid) were prepared from Merck (Darmstadt, Germany).
The preparation of hydroalcoholic extract
The roots of R. turkestanicum were acquired from Mashhad, Khorasan Razavi Province, Iran, and it was verified by Joharchi at Ferdowsi University of Mashhad (herbarium number: 21377). Briefly, the dried powder of the roots (50 g) were separated with 70% ethanol in a Soxhlet apparatus for 48 hours. The obtained hydroalcoholic extract was dried in a water bath and then kept at -18°C in a freezer. The yield of the extract was 19% (w/w).
Cell culture and treatments
SH-SY5Y was bought from the National Cell Bank of Iran (NCBI, Tehran, Iran). SH-SY5Y cells were cultured in an incubator in a humidified atmosphere containing CO2 (5%) and then passaged at 80% confluence. Pretreatments were done by different concentrations of R. turkestanicum extract (12.5 to 100 µg/mL), and then 6-OHDA was added (0 to 85 µg/mL).
Measurement of cytotoxicity
SH-SY5Y cells were cultured at a density of 104 cells/well onto flat-bottomed 96-well culture plates. Then, the cells were exposed to a toxic agent (0 to 85 µg/mL) for 24 hours. The MTT assay evaluated cytotoxicity, and the final concentration of 6-OHDA was calculated based on the IC50. Subsequently, the cells were treated with 12.5 to 100 µg/mL of the extract for 2 hours and then exposed to 6-OHDA (final concentration 42.5 µg/mL) for 24 hours to determine the inhibitory effect of R. turkestanicum on 6-OHDA-induced cytotoxicity. Then, MTT solution (0.5 mg/mL) was added to the cells for 4 hours. The absorbance was read using a microplate reader (Start Fax-2100, UK) at 545 nm (630 nm as a reference) (16). All the treatments were done in triplicates.
Measurement of ROS amount
The ROS level was measured by using the 2,7-dichlorofluorescein diacetate (DCFH-DA) method (7). Briefly, after exposure of the cells to various concentrations of the extract (12.5 to 100 µg/mL) for 24 hours, 6-OHDA was added to the cells at the IC50 dose of (42.5 µg/mL). After 24 hours of incubation, the upper-medium was separated, and the cells were incubated with DCFH-DA (10 µM) for 30 min at 37°C in the dark. Next, the cells were washed with PBS, and the fluorescence intensity of DCF was determined using the FACScan fluorescence plate reader (Becton Dickinson, San Jose, USA) with an excitation/emission of 485/535 nm. All the treatments were done in triplicates.
Lipid peroxidation assay
The MDA level was measured as the end product of lipid peroxidation (17). SH-SY5Y cells were pretreated with 25, 50, and 100 μg/mL of the extract for 24 hours before exposure to 42.5 µg/mL 6-OHDA, followed by an additional 24 hours of incubation. Next, the centrifugation of the cells was done at 13,000 g at 4°C for 30 min. Then, 400 µL of TCA (15%) and 800 µL of TBA (0.7%) were added to 500 µL of the cell samples. The mixture was vortexed and then heated for 40 min, and then 200 µL of the sample was transferred to a 96-well plate, and the fluorescence intensity was read with excitation/emission of 480/530 nm. All the treatments were performed in triplicates.
Flow cytometry analysis of apoptosis
The sub-G1 peak of the treated cells was detected by PI staining and flow cytometry to evaluate the number of apoptotic cells (18). SH-SY5Y cells were pretreated with 25, 50, and 100 μg/mL of the extract for 24 hours before exposure to 42.5 µg/mL 6-OHDA, followed by an additional 24 hours of incubation. Next, the cells were incubated at 37°C in the dark with 400 μL of hypotonic buffer (50 μg/mL PI in 0.1% sodium citrate and 0.1% Triton X-100) for 30 min. Then, cell cycle distribution was analyzed from 104 cells using a BD Facscalibur™ flow cytometer (Becton Dickinson, Mountain View, CA, USA).Afterward, the cell cycle analysis of flow cytometry data was done utilizing the software FlowJo® vX.0.7 (Tree Star, Ashland, OR, USA) (19, 20). All the treatments were done in triplicates.
Measurement of GSH level
The content of the sulfhydryl group was evaluated via the formation of yellow color in the presence of DTNB [5, 5′-Dithiobis (2-nitrobenzoic acid)]. In this regard, phosphate-buffered saline was applied to wash the cells, and then 15% TCA was added to extract GSH from the cells. After centrifugation at 14,000 rpm for 10 min, the denatured proteins were isolated. In brief, TCA extract (500 mL) was mixed with 1 mL of a reaction mixture containing 0.1 M sodium phosphate buffer (pH 7.5) and 0.6 mM DTNB. A spectrophotometer was used to measure the absorbance at 412 nm for 2 min.
Measurement of SOD activity
The effective doses of the extract were added to the cells and then incubated with 250 μM 6-OHDA for 24 hours to determine SOD activity. The activity was measured by spectrophotometry (at 405 nm) based on the prevention of pyrogallol autoxidation as described previously. The autoxidation of pyrogallol in tris-cacodylic acid buffer (0.05 M, pH 8.2) was measured.
Statistical analysis
All data were displayed as the mean±SEM of at least three independent tests. The data were exposed to statistical analysis via one-way ANOVA followed by Dunnett's test with GraphPad Prism® 7.01 software (GraphPad Software, Inc., San Diego, CA). Mean values were considered to be statistically significant at p<0.05.
R. turkestanicum extract attenuates 6-OHDA-induced cytotoxicity
As demonstrated in Fig. 1A, the treatment of SH-SY5Y cells with 6-OHDA for 24 hours remarkably reduced the cell viability concentration dependently (p<0.05 and p<0.001). Moreover, the IC50 for 6-OHDA was about 42.5 µg/mL. Therefore, the treatment of 42.5 µg/mL 6-OHDA for 24 hours induced SH-SY5Y cellular toxicity in sequential experiments. To evaluate the cytotoxicity of R. turkestanicum, we subjected SH-SY5Y cells to 6.25–200 µg/mL concentrations of R. turkestanicum hydroalcoholic extract for 24 hours, as shown in Figure. 1B. The results showed that R. turkestanicum extract in concentrations of 6.25 to 200 µg/mL did not induce any apparent cytotoxicity (p>0.05, Figure. 1B).
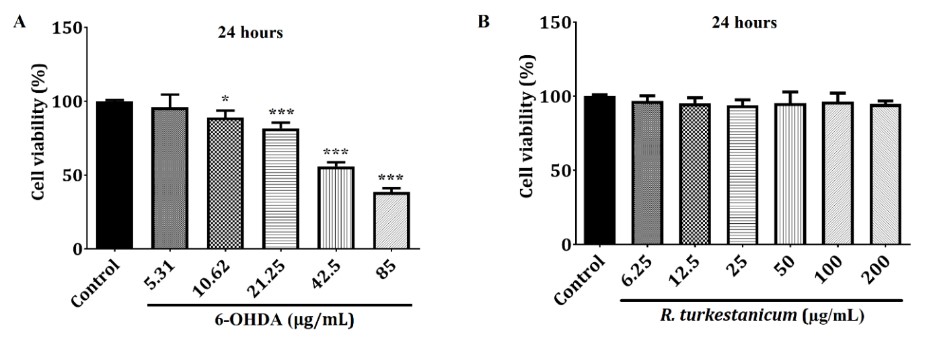
Figure 1. 6-OHDA (A) and R. turkestanicum (B) cytotoxicity in SH-SY5Y cells. The MTT assay evaluated R. turkestanicum and 6-OHDA cytotoxicity. The IC50 of 6-OHDA was determined at about 42.5 μg/mL for 24 hours of treatment (A). R. turkestanicum had no significant change in the survival of SH-SY5Y cells after 48 hours (B). Each column represents the mean ± SEM for each group. *P<0.05 and ***P<0.001 versus the control group (n=8).
Based on these data, the cells were pretreated with 12.5, 25, 50, and 100 µg/mL of the extract for 24 hours before exposure to 42.5 µg/mL 6-OHDA, followed by an additional 24 hours of incubation to test whether R. turkestanicum treatment increased cell survival decreased by 6-OHDA. As a result, the treatment of SH-SY5Y cells with 6-OHDA markedly reduced cell viability compared to the control group (p<0.001) for 24 hours. Interestingly, the pretreatment with R. turkestanicum extract (12.5, 25, 50, and 100 µg/mL) significantly attenuated the cytotoxic effects induced by 6-OHDA compared to the 6-OHDA-treated cells (Figure 2, p<0.001).
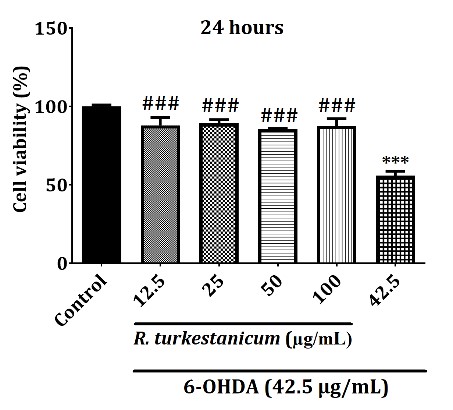
Figure 2. R. turkestanicum extract attenuated 6-OHDA-induced cytotoxicity in SH-SY5Y cells. SH-SY5Y cells were incubated with R. turkestanicum (12.5, 25, 50, and 100 µg/mL) for 24 hours and stimulated with 6-OHDA (42.5 µg/mL) for 24 hours. Cytotoxicity was determined by utilizing the MTT assay. The tests were repeated three times independently, with one representative result shown. The data are expressed as the mean ± SEM of five determinations in a representative experiment. ***p<0.001 versus the control group; ###p<0.001 versus the 6-OHDA-treated cells.
R. turkestanicum extract diminishes ROS and MDA production induced by 6-OHDA
To determine whether R. turkestanicum extract could suppress 6-OHDA-initiated oxidative stress, we measured the ROS and MDA levels. As shown in Fig. 3, after treatment of the cells with 42.5 µg/mL 6-OHDA, the ROS and MDA levels were substantially expanded compared to the control group (p<0.001), suggesting that 6-OHDA could incite oxidative stress in SH-SY5Y cells.
Interestingly, pretreatment with R. turkestanicum extract (6.25, 12.5, 25, and 50 µg/mL) for 24 hours, followed by exposure to 42.5 µg/mL 6-OHDA for 24 hours, decreased the ROS level significantly compared to the 6-OHDA-treated cells (Fig. 3A, p<0.05 and p<0.01).
Also, twenty-four hours after incubation, the level of lipid peroxidation was examined by measuring the MDA level. In Figure. 3B, the exposure of SH-SY5Y cells to 6-OHDA resulted in a significant increase in the MDA level (192.72%, p<0.001) compared to control cells cultured in the absence of 6-OHDA (100%). The MDA content was significantly diminished in cells pretreated with 25 µg/mL (121%, p<0.001), 50 µg/mL (130%, p<0.001), and 100 µg/mL (121%, p<0.001) of R. turkestanicum extract compared to the 6-OHDA-treated cells.
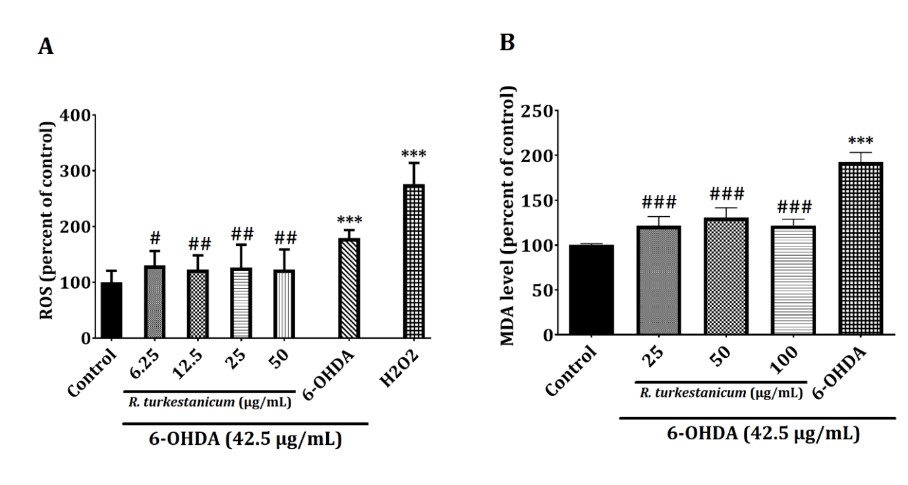
Figure 3. (A) R. turkestanicum extract suppressed the 6-OHDA-induced ROS level, which induced apoptosis in SH-SY5Y cells. SH-SY5Y cells were incubated with R. turkestanicum (6.25, 12.5, 25, and 50 µg/mL) for 24 hours and stimulated with H2O2 (200 µM) for 2 hours and 6-OHDA (42.5 µg/mL) for 24 hours. The ROS level was determined to utilize a fluorescence probe, DCFH-DA. The tests were repeated three times independently, with one representative result demonstrated. The data are expressed as the mean ± SEM of five determinations in a representative experiment. ***p<0.001 versus the control group; ##p<0.01 and #p<0.05 versus the 6-OHDA-treated cells. (B) R. turkestanicum extract suppressed 6-OHDA-induced MDA production, which induced oxidative damage in SH-SY5Y cells. SH-SY5Y cells were incubated with R. turkestanicum (25, 50, and 100 µg/mL) for 24 hours and stimulated with 6-OHDA (42.5 µg/mL) for 24 hours. The tests were repeated three times independently, with one representative result shown. The data are expressed as the mean ± SEM of five determinations in a representative experiment. ***p<0.001 versus the control group; ###p<0.001 versus the 6-OHDA-treated cells.
R. turkestanicum extract suppresses 6-OHDA-induced apoptosis
As shown in Figure 4, following 24 hours of incubation with 42.5 µg/mL 6-OHDA, the outcomes indicated that in 6-OHDA treatment alone, the percentage of apoptotic cells (67.0 %, p<0.001) was expanded markedly compared with the normal control (11.6 %). However, it respectively dropped to 53.1, 48.6, and 28.1% after 25, 50, and 100 µg/mL of R. turkestanicum extract treatments.
R. turkestanicum extract increases GSH levels and SOD activity
We measured SOD activity and the GSH level to determine whether R. turkestanicum extract could mediate the 6-OHDA-reduced GSH level and SOD activity. As shown in Fig. 5, after treatment of the cells with 42.5 µg/mL 6-OHDA, the GSH and SOD levels substantially decreased compared to the control group (p<0.01 and p<0.001, respectively). Interestingly, pretreatment with R. turkestanicum extract (25, 50, and 100 µg/mL) for 24 hours, followed by exposure to 42.5 µg/mL 6-OHDA for 24 hours, increased the GSH level significantly compared to the 6-OHDA-treated cells (Fig. 5A, p<0.001).
Also, SOD activity was measured 24 hours after incubation. In Fig. 5B, the exposure of SH-SY5Y cells to 6-OHDA resulted in a significant decrease in SOD activity (p<0.001) compared to control cells cultured in the absence of 6-OHDA (100%). The content of SOD activity significantly increased in cells pretreated with 25, 50, and 100 µg/mL of R. turkestanicum extract compared to the 6-OHDA-treated cells.
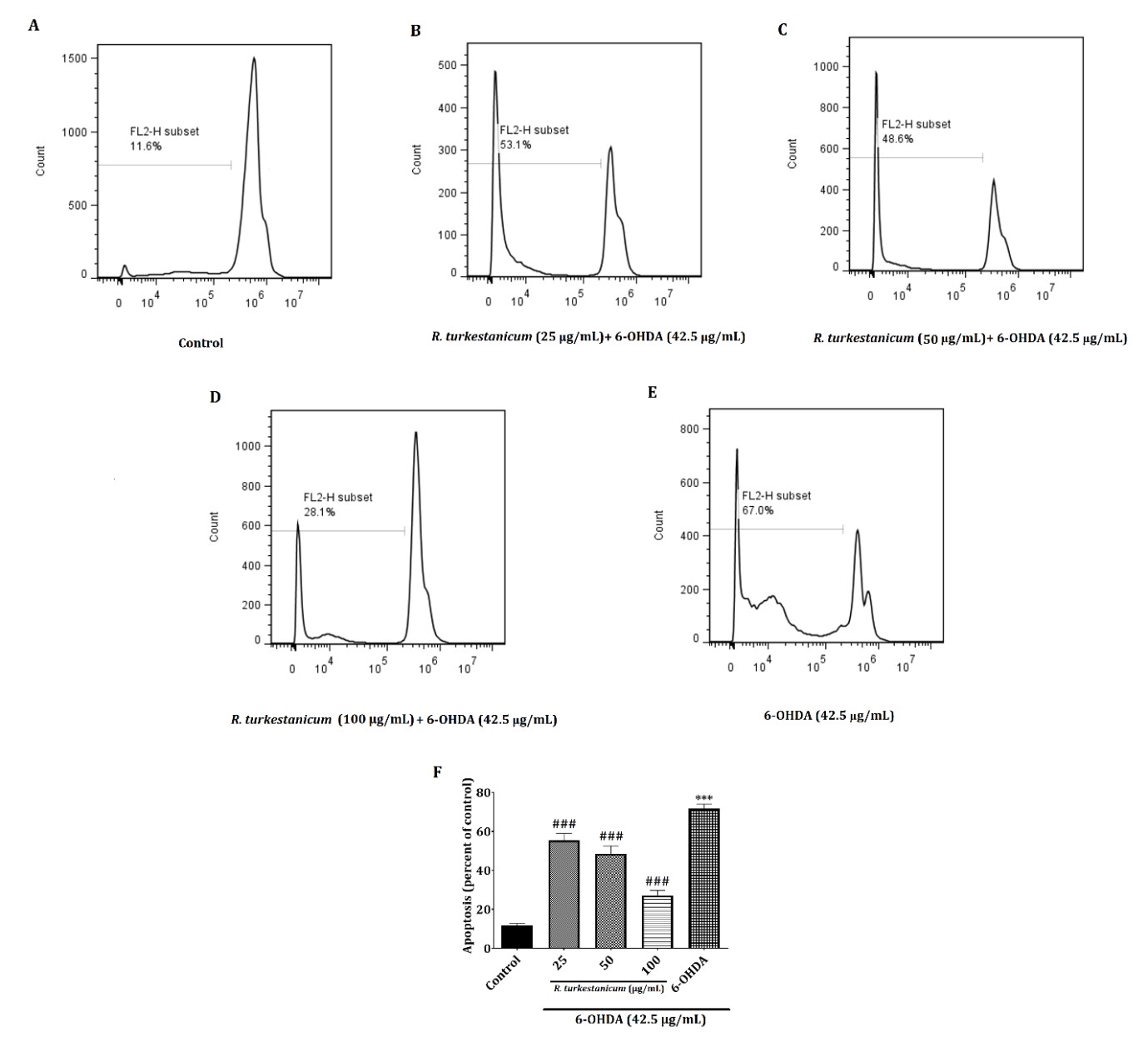
Figure 4. The effect of R. turkestanicum extract on 6-OHDA-induced apoptosis in SH-SY5Y cells. SH-SY5Y cells were incubated with different concentrations of R. turkestanicum (25, 50, and 100 µg/mL) and 6-OHDA (42.5 µg/mL) for 24 hours. Then, the apoptosis rate was measured with a PI staining. Values are shown as the mean ± SEM. The tests were repeated three times independently, with one representative result shown. ***p<0.001 versus the control; ###p<0.001 versus the 6-OHDA-treated cells.
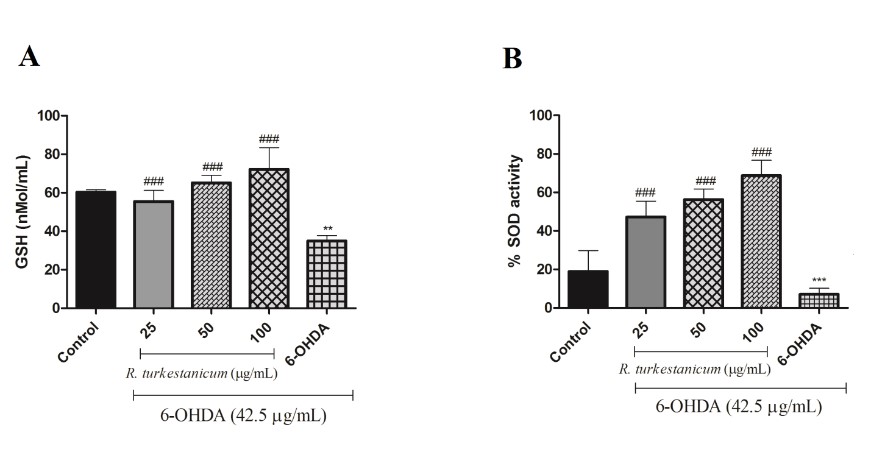
Figure 5. (A) R. turkestanicum extract increased the 6-OHDA-reduced GSH level. SH-SY5Y cells were incubated with R. turkestanicum (25, 50, and 100 µg/mL) for 24 hours and stimulated with 6-OHDA (42.5 µg/mL) for 24 hours. Data are expressed as the mean ± SEM of five determinations in a representative experiment. **p<0.01 versus the control group; ###p<0.001 versus the 6-OHDA-treated cells. (B) R. turkestanicum extract increased 6-OHDA-reduced SOD activity. SH-SY5Y cells were incubated with R. turkestanicum (25, 50, and 100 µg/mL) for 24 hours and stimulated with 6-OHDA (42.5 µg/mL) for 24 hours. The tests were repeated three times independently, with one representative result shown. The data are expressed as the mean ± SEM of five determinations in a representative experiment. ***p<0.001 versus the control group; ###p<0.001 versus the 6-OHDA-treated cells.
Discussion
The neuropathological and biochemical features of PD can be presented by 6-OHDA, a neurotoxic agent frequently used to explore molecular pathway patterns involved in cytotoxicity, apoptosis, and oxidative stress in human neuroblastoma SH-SY5Y cells. 6-OHDA is oxidized under physiological circumstances, generating a variety of ROS, including p-quinines and hydrogen peroxides, causing damage to the DNA, lipid peroxidation, and cell death (21-23). The present study showed that R. turkestanicum effectively inhibited 6-OHDA-induced neuronal toxicity in SH-SY5Y cells.
While numerous studies have reported that medicinal herbs can preserve cells against oxidative stress-induced toxicity (6, 7, 24-28), it remains unclear whether R. turkestanicum, one of the promising medicinal plants, can protect neuronal cells against oxidative stress-incited toxicity. R. turkestanicum, as a natural medicine utilized in traditional medicine, has polyphenol ingredients like anthraquinones, stilbenes, anthocyanins, and flavonols (12). The majority of its components have antioxidant activities against toxicological conditions. Studies have indicated that this potential plant is utilized to treat diabetes, hypertension, and cancer (10, 13). Hosseini et al. demonstrated the protective effects of R. turkestanicum against doxorubicin in cardiomyocytes cells (10), although its protective effects on neuronal toxicity have not been elucidated.
Oxidative stress, as a fundamental reason for cell damage in various debilitating diseases, including neurodegenerative disorders, refers to the imbalance between the formation and clearance of free radical species within the cells. Evidence suggests that 6-OHDA-induced ROS generation plays a primary role in the induction of neuronal damage and apoptosis (29). Hence, evacuation of excess ROS or concealment of the ROS level might be valuable in preventing oxidative neuronal cell death (30, 31). The defensive impacts of R. turkestanicum on the cytotoxic activity induced by 6-OHDA were examined to determine the mechanism by pretreatment with R. turkestanicum against 6-OHDA-induced injury. It was found that cytoprotective action was of particular interest in oxidative stress (32). In the present study, we found that R. turkestanicum significantly mitigated 6-OHDA-induced ROS generation and cytotoxicity in SH-SY5Y cells.
Moreover, MDA reflects the oxidative injury of the plasma membrane, and the resulting TBA reactive substances are related to oxidant stress and lipid peroxidation (33). Hosseini and his colleagues showed that R. turkestanicum (12-100 µg/mL) reduced H2O2-induced oxidative stress by reducing MDA levels (34). We showed that MDA levels decreased after treatment with R. turkestanicum. Also, R. turkestanicum extract significantly reduced the apoptosis rate due to 6-OHDA concentration-dependently.
Different studies have reported that 6-OHDA utilizes its toxic effects by producing superoxide anions and hydrogen peroxide. The SOD enzyme converts the superoxide radicals to hydrogen peroxide (35-37). Our results showed that 6-OHDA significantly reduced the GSH level and SOD activity while R. turkestanicum extract increased these parameters.
The study's results prove that cell oxidative injury plays an essential role in the toxicity of 6-OHDA and that pretreatment with R. turkestanicum can attenuate these changes. This study indicated that R. turkestanicum significantly diminished 6-OHDA-induced cytotoxicity, apoptosis, and oxidative stress compared to the 6-OHDA treated-cells. According to the presence of tannins and phenolic compounds (gallic acid) in R. turkestanicum (13, 38), the protective effect of R. turkestanicum is probably related to its components and other ingredients with antioxidant activities.
Conclusion
The current study offers the first evidence of the protective impact of R. turkestanicum on neuronal toxicity. The present outcomes showed that R. turkestanicum, a potential medicinal herb, markedly suppressed 6-OHDA-induced neuronal toxicity in human neuroblastoma SH-SY5Y cells, presumably through attenuating ROS, generating MDA, apoptosis, and increasing SOD activity and the GSH level. Albeit, further broader examinations of in vivo models of neurodegenerative diseases are essential to recogn.
Acknowledgements
None.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Received: 2021/05/11 | Accepted: 2022/07/13 | Published: 2022/10/10
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy