BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://journal.zums.ac.ir/article-1-7434-en.html


 , Mohammadreza Fathi2 , Ali Khodadadi3 , Ahmad Shamsizadeh4 , Ata A Ghadiri1 , Pegah Ghandil *5
, Mohammadreza Fathi2 , Ali Khodadadi3 , Ahmad Shamsizadeh4 , Ata A Ghadiri1 , Pegah Ghandil *5
2- Department of Pediatrics, Faculty of Medicine, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
3- Department of Immunology, School of Medicine, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
4- Infectious and Tropical Diseases Research Center, Health Research Institute, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
5- Cellular and Molecular Research Center, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran & Diabetes Research Center, Health Research Institute, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran ,
Familial Mediterranean Fever (FMF) is the commonest monogenic auto-inflammatory disease. The clinical features include recurrent self-limiting attacks of fever, peritonitis, pleuritis, arthritis, and cutaneous rashes. Amyloidosis is the most significant complication of FMF, leading to kidney failure (1). The first attack and symptoms typically occur in early childhood. FMF is mainly observed in individuals of Mediterranean descent, including Turks, Greeks, Jews, Arabs, Armenians, and Persians. The prevalence of FMF in endemic countries is about 1:500 to 1:1000, and it is rare in other ethnic populations (2). The precise prevalence of FMF in Iran is yet to be determined, highlighting the need for further research, but the estimated carrier rate is 27.4 % (3).
Serum amyloid-A protein (SAA), haptoglobin, ceruloplasmin, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), fibrinogen, leukocyte count, and some cytokines are acute phase reactants that rise in the blood during attack periods. These findings may help support the diagnosis or rule out other causes (4).
The diagnosis of FMF, especially in endemic regions, is generally clinical, using the Tel Hashomer criteria (5). Genetic evaluations are crucial for confirming the diagnosis in patients with the atypical phenotype of FMF. However, 10% to 20% of patients showing typical clinical manifestations lack a mutation in the MEFV gene (6).
Mutations in the Mediterranean fever gene (MEFV) cause FMF. MEFV is located on chromosome 16p13.3, consists of 10 exons, and encodes the protein pyrin/marnosterin. Pyrin is expressed mainly in monocytes, neutrophils, eosinophils, and fibroblasts, which play an essential role in inflammatory responses by regulating caspase-1 activation and processing of mature IL-1β (2).
Mutations in the MEFV gene cause abnormal synthesis of pyrin proteins. In mutated conditions, inflammatory regulation does not occur correctly. Colchicine is the therapy of choice for patients with FMF as it inhibits attacks and the progression of amyloidosis. Interleukin-1 (IL-1) inhibitors are the first-line alternatives in colchicine non-responder/intolerant FMF patients (7).
So far, the Infevers database (8) has recorded 399 mutations and polymorphisms in the MEFV gene, mostly located in exons 2, 10, 5, 3, and 1, with over 96% being substitutions. The distribution of MEFV gene variants differs across different ethnic groups. In Iranian patients with FMF, the most prevalent mutations are p.M694V, p.E148Q, p.V726A, p.R761H, p.M680I (both c.2040G>C and c.2040G>A), and p.M694I (9).
So far, several studies from different regions of Iran have examined the spectrum of MEFV gene variants in patients with FMF. Limited studies have been done in southwest Iran, underscoring the need for additional research in this region. In the present study, we sequenced all 10 exons of the MEFV gene in 40 unrelated pediatric patients with clinical suspicion of FMF in the Southwest of Iran. We aimed to explore all mutations in all exons of the MEFV gene and compare them with the spectrum of MEFV gene variants in other regions of Iran and other countries, considering the patients' clinical phenotypes.
2.1 Patients
Under the supervision of a pediatric rheumatology specialist, we chose 40 unrelated pediatric patients suspected of having FMF. Between April 2020 and July 2024, these patients were referred to the pediatric rheumatology clinic of Abuzar Children's Hospital and some other referral medical centers in Khuzestan province. Abuzar Children's Hospital, affiliated with Ahvaz Jundishapur University of Medical Sciences, is one of southwestern Iran's main pediatric referral hospitals. Patients and their families provided written informed consent, and the Ethics Committee of Ahvaz Jundishapur University of Medical Sciences approved the study (IRAJUMS.MEDICINE.REC.1399.003).
The Tel Hashomer criteria were used for the clinical diagnosis of FMF. Typical clinical findings were recurrent episodes of fever, abdominal pain, vomiting, myalgia, joint pain, and chest pain. Most of the patients had parents who had consanguineous marriages, and all of them were from the southwest of Iran, representing a variety of ethnic backgrounds, such as Fars, Arabs, and Lors. The data concerning the clinical findings during attacks, laboratory tests, and familial history of FMF were collected. In addition, parents of patients with rare MEFV gene mutations were investigated for the mutations detected in probands.
2.2 Sequencing and mutation detection
DNA extraction from blood samples containing EDTA anticoagulant was performed using the DNA isolation kit (YEKTA TAJHIZ: cat. No: FABGK001). NanoDrop spectrophotometry (ND-2000, Thermo Scientific, Wilmington, USA) quantified the concentration and purity of the extracted DNA samples. For further tests, the DNA samples were stored at -20°C. Polymerase chain reaction (PCR) amplification was performed for all 10 exons of the MEFV gene with conditions and primers described previously (10). The quality of PCR products was assessed using 1.5% agarose gel electrophoresis. All PCR products were sequenced using the Sanger sequencing method, carried out using the Big Dye Terminator reaction kit and an ABI Prism 3700 apparatus (Applied Biosystems).
Chromas Pro v2.1.3 was used to analyze the Sanger sequencing results, with online bioinformatics tools used to align the results with reference sequences. Genomic and amino acid sequences of the MEFV gene were collected from Ensembl: ENST00000219596 (https://www.ensembl.org/index.html) and the 'Nucleotide Blast' Alignment Program at http://blast.ncbi.nlm.nih.gov/Blast.cgi.
2.3 In silico analysis
In this study, various in silico tools were employed to predict the pathogenicity and the effect of the p.S339F mutation on the structure and function of the pyrin protein. These tools included PolyPhen (http://genetics.bwh.harvard.edu/pph2), Mutation Taster (http://www.mutationtaster.org/), and CADD (http://cadd.gs.washington.edu/) to determine the pathogenicity of the mutation identified. Furthermore, the stability of the mutant protein in comparison to the wild-type protein was examined using the I-Mutant 2.0 site (https://folding.biofold.org/cgi-bin/i-mutant2.0.cgi).
3.1 Clinical Findings
The present study examined 40 unrelated pediatric patients, including 22 boys (55%) and 18 girls (45%). The age range of the patients was 1-18 years. All patients revealed the first clinical symptoms before the age of 10. The studied patients had the severe clinical form of the disease, such as earlier age of onset, severe attacks requiring bed rest, and long-term persistence. All patients with mutations in the MEFV gene had consanguineous parents, among whom 37.5% were first cousins, while the rest were third and 4th degree. In 43.75% of patients, a positive family history of FMF was present, particularly among close relatives.
The main clinical findings were fever (100%), abdominal pain (100%), vomiting (68.75%), myalgia (56.25%), joint pain (43.75%), chest pain (37.5%), and seizures (6.25%) (Table 1). There were no signs or symptoms of amyloidosis in our patients. Tonsillectomy was detected in one patient. In addition, oral leukoplakia was reported in two patients. In some patients during the past attacks, laboratory data indicated that the WBC count, CRP, and ESR levels were significantly higher than those in periods without attack (Table 1). All patients were treated with colchicine during attack-free periods.
Table 1. Demographic, Clinical features and Genetic findings of patients
| Clinical Characteristic | Familial History of FMF | Genotype | Mutation | Age at onset(y) | Age( y) /gender | FMF Patients | ||||||||
| ESR/CRP↑ | S | V | M | J | C | A | F | |||||||
| - | - | + | + | - | - | + | + | + | HOM | A744S | 3 | 11/F | P1 | |
| - | - | + | + | + | + | + | + | + | C.HET | M694V/M694I | 5 | 18/F | P2 | |
| + | + | - | - | - | - | + | + | + | C.HET | V726A/E148Q | 4 | 14/M | P3 | |
| - | - | - | + | - | - | + | + | - | C.HET | A744S/E148Q | 1 | 17/F | P4 | |
| + | - | + | + | + | - | + | + | - | C.HET | M680I/E148Q | 1 | 11/M | P5 | |
| - | - | + | - | + | + | + | + | + | C.HET | M694I/E148Q | 2 | 5/F | P6 | |
| + | - | - | - | + | + | + | + | - | HOM | M694V | 4 | 8/M | P7 | |
| + | - | + | + | + | - | + | + | + | C.HET | M694V/R202Q | 2 | 4/F | P8 | |
| + | - | - | + | - | - | + | + | - | C.HET | S339F/E148Q | 6 | 13/M | P9 | |
| - | - | + | - | - | + | + | + | - | HET | M694I/WT | 3 | 5/F | P10 | |
| + | - | + | + | - | - | + | + | - | C.HET | R202Q/E148Q | 4 | 7/F | P11 | |
| + | - | + | - | + | - | + | + | + | C.HET | M694V/R202Q | 1 | 2/M | P12 | |
| - | - | - | - | - | + | + | + | - | C.HET | R202Q/E148Q | 3 | 10/M | P13 | |
| - | - | + | + | - | - | + | + | - | C.HET | V726A/R202Q | 6 | 8/M | P14 | |
| - | - | + | - | - | - | + | + | - | C.HET | R202Q/E148Q | 2 | 13/F | P15 | |
| + | - | + | + | + | + | + | + | + | Complex | M694V/M694I/R202Q | 2 | 17/F | P16 | |
Abbreviations: HET: Heterozygous, HOM: Homozygous, C.HET: Compound Heterozygous, WT: Wild Type
+: present, −: absent, y: years, A: Abdominal pain, F: Fever, C: Chest pain, V: vomiting, J: Joint pain
↑: increased, S: seizures, M: Myalgia, P: patient.
3.2 Genetic Findings
The spectrum of MEFV gene mutations for the studied patients is listed in Table 1. MEFV mutations were found in 40% (n=16) of patients, while 60% (n=24) had no mutations. We detected eight mutations: two in exon 2, one in exon 3, and five in exon 10. No mutations were found in the other seven exons. Six out of eight detected mutations were among common mutations, including p.E148Q, p.M694V, p.M694I, p.A744S, p.V726A, and p.M680I (c.2082G>A). We also identified an uncommon mutation p.R202Q (c.605G>A, rs224222) located in exon 2 (Figure 1). This mutation was identified in seven patients, six of whom had a compound heterozygous form (p.M694V/p.R202Q in P8 and P12, p.R202Q/p.E148Q in P11, P13, and P15, and p.V726A/p.R202Q in P14), and one had a complex genotype (p.M694V/p.M694I/p.R202Q in P16) (Table 1).
Meanwhile, we detected a rare missense mutation (p.Ser339Phe, c.1016C>T, rs104895157) located in exon 3 (Figure 2) in P9 as a compound heterozygote with p.E148Q (Table 1). This variant was reported in the ClinVar database (Variation ID: 36497).
Based on the Mutation Taster server, this substitution mutation (c.1016C>T) was probably pathogenic for FMF. It was located on the coding sequence, which changed the amino acid serine to phenylalanine at position 339 (p. S339F). Furthermore, with a score of 0.989 (sensitivity: 0.72; specificity: 0.97), the PolyPhen server predicted that this mutation was probably damaging. Also, CADD predicted this mutation to be deleterious (Score: 23.1). Moreover, for this mutation, the free energy change (ΔΔG) value determined by the I-Mutant server was -0.24 kcal/mol. Thus, the structural stability of this mutant protein diminished.
Regarding the analysis of the P9 family, the father was heterozygous for p.E148Q, while his mother had p.S339F with heterozygote states. His parents were healthy in terms of FMF and had no symptoms.
Among 16 patients in whom mutations were detected, 12 (75%) were compound heterozygous, 2 (12.5%) were homozygous, 1 (6.25%) was heterozygous, and 1 (6.25%) had a complex genotype. Concerning the detected mutant alleles, the most frequently mutant allele was p.E148Q (50%), followed by p.R202Q (43.75%), p.M694V (37.5%), p.M694I (25%), p.A744S (18.75%), p.V726A (12.5%), p.M680I (6.25%), and p.S339F (6.25%). Also, the most common genotype was p.R202Q/p.E148 (18.75%), based on the data reported in Table 1.
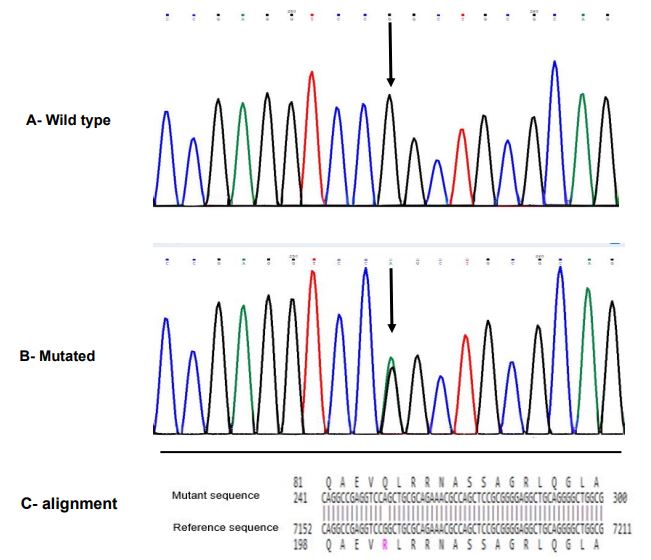
Figure 1. Sanger sequencing chromatogram and alignment to the reference sequence. A, Wild-type sequence with the normal homozygous state; B, p.R202Q mutation in exon 2 of the MEFV gene (NM_000243.3: c.605G>A (p.Arg202Gln)) with heterozygous state; C, BLAST sequence alignment between mutant sequence and reference sequence gene(MEFV)
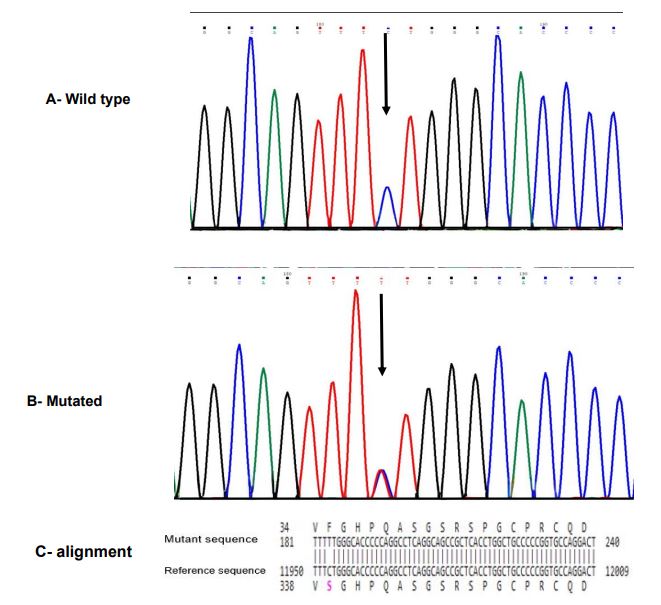
Figure 2. Sanger sequencing chromatogram and alignment to the reference sequence. A; Wild-type sequence with the normal homozygous state; B, p.S339F mutation in exon 3 of the MEFV gene (NM_000243.3:c.1016C>T (p.Ser339Phe)) with heterozygous state; C, BLAST sequence alignment between mutant sequence and reference sequence gene(MEFV)
Discussion
To confirm the diagnosis of FMF disease, molecular investigation of the MEFV gene and identification of relevant mutations are necessary. This study confirmed FMF in 16 out of 40 unrelated pediatric patients. We identified six common mutations, including p.E148Q, p.M694V, p.M694I, p.A744S, p.V726A, and p.M680I (c.2082G>A). We also identified one uncommon mutation (p.R202Q) and one rare mutation (p.S339F) in our patients. Although several reports from Iran described p.M694V as the most common mutation (9), p.E148Q was the most prevalent mutation in our patients (50%). Similar to our research, the studies of Haghighat et al., conducted on southwest Iranian patients in 2017 (n=20), and Malek et al., performed on northeast Iranian patients in 2023 (n=29), found that the most frequent mutation was p.E148Q, with a frequency of 25% and 41.3%, respectively (11, 12).
This study detected the rare mutation p.S339F, representing the first case reported in Iranian populations. Our study identified this rare mutation in P9 as a compound heterozygote with p.E148Q (a 13-year-old boy). When he was six years old, he had recurrent episodes of abdominal pain, fever, and myalgia. The patient responded to treatment with colchicine. This mutation was registered for the first time in 2006 from Turkey on the Infever database (8). Thus far, it has been reported in only a few patients worldwide. In the two separate studies, one including 15 patients by Stella et al. in 2019 from Italy and the other including 56 subjects by Cetin et al. in 2020 from Turkey, the mutation p.S339F was reported in combination with one or two other mutations in the MEFV gene (p.A744S/p.S339F/p.Asn256Argfs*70 and p.S339F/p.R202Q). However, the detailed clinical features of the reported patients were unavailable (13, 14).
This rare mutation, p.S339F, is in exon 3 of the MEFV gene. This exon encodes the B-box domain, which interacts intramolecularly with the PYD (pyrin domain) to maintain pyrin in a repressed state. Therefore, it can be assumed that mutations in this region change the auto-inhibitory role of the domain (15). In the study by Stella et al. (13), they confirmed the pathogenicity of this mutation using different in silico tools. However, we confirmed, for the first time, its pathogenicity using the I-Mutant site. The I-Mutant website indicates that this substitution mutation alters the protein's free energy, thereby causing instability in its structure (16).
We identified an uncommon mutation, p.R202Q, in seven of 16 patients (43.75%). The p.R202Q variant was initially reported as a polymorphism (17). A close association was identified between this variation and FMF disease in the following years, which can be regarded as a mutation. Recent studies have shown its importance as a risk factor for the development of amyloidosis (18, 19). In the studies of Altamimi et al. in 2023 (n=132) and Capraz et al. in 2023 (n=1570) from Turkey, the most prevalent mutation was p.R202Q, with a frequency of 53.8% and 43.5%, respectively (20, 21). Although the p.R202Q mutation is uncommon in Iran (9), it was the second most prevalent mutation in the present study, with an allelic frequency of 43.75%. This mutation was observed in the compound heterozygous state (combination with p.E148Q, p.M694V, and p.V726A mutations) and complex genotype (p.M694V/p.M694I/p.R202Q). This rare complex genotype (p.M694V/p.M694I/p.R202Q) had not previously been reported in Iranian studies but was reported by Arpaci et al. in 2021 from Turkey (18).
Similar to our results, studies conducted by Beheshtian et al. in 2016 (n=951) and Farahzadi et al. in 2021 (n=252) identified genotypes p.R202Q/p.E148Q and p.M694V/p.R202Q in suspected FMF patients (3, 22). In a recent study by Nourbakhsh et al. in the southwest of Iran (n=13), p.R202Q was the most prevalent mutation, with a frequency of 38.46% (23). Differences in the frequency of the p.R202Q mutation in different populations, including Iran, might arise from socio-demographic characteristics, ethnic differences (considering the multiethnic nature of the Iranian population), and the number of studied patients. These results revealed that further studies are necessary to understand the prevalence and implications of the p.R202Q mutation in Iran.
To date, five mutations (p.M694V, p.M694L, p.M694del, p.M694I, and p.M694K) have been detected at codon 694 of the MEFV gene (Infevers, 2024). The methionine residues in codons 694 and 680 play a critical role in the function of pyrin. Thus, mutations in these codons can cause severe symptoms. In particular, the p.M694V mutation causes the most complicated form of FMF, and its homozygosity penetration is considerably high (99%) (24). As expected, according to the Tel-Hashomer criteria, our patients with mutations in these codons had a severe phenotype.
The pattern of inheritance for FMF is autosomal recessive, with a dominant inheritance hypothesis supported by numerous studies (OMIM 134610). P10 showed this pattern in our research. Although the whole gene was analyzed, we found only one mutation in this patient. This patient had a p.M694I (c.2040G>A) mutation and suffered from typical symptoms of FMF. Some studies have reported a dominant inheritance pattern for this mutation (25, 26).
Similar to other studies (6, 27), in spite of the clinical diagnosis, no mutation was observed in the coding and splicing regions of the MEFV gene in 24 out of the 40 studied patients. Genetic factors influence the occurrence of FMF, but it is also susceptible to various other influences, such as mutations in other auto-inflammatory genes, epigenetic or post-translational changes, environmental factors, and hidden biomarkers (28). It is important to note that PFAPA syndrome can sometimes present with FMF-like symptoms, and these patients respond to colchicine (29, 30). This potential overlap underscores the need for careful and thorough differential diagnosis. Meanwhile, this study did not cover the MEFV gene intronic regions.
FMF diagnosis is based on clinical manifestations as well as genetic testing and is supported by ethnic origin and family history. All parents of the patients in whom the mutation was detected had consanguineous marriages. Specifically, 37.5% were first cousins and the rest were distant relatives. In 43.75% of patients, a positive family history of FMF was present, particularly among close relatives. In the 2019 study by Rostamizadeh et al. on Azari-Turkish FMF patients, the ratio of parenteral consanguinity and family history of FMF was found to be 24.2% and 20%, respectively (6).
Regarding the clinical presentation of FMF, fever and abdominal pain are the most common complaints for all ethnicities (31). The frequency of clinical manifestations varies across populations. In the present study, the most prevalent clinical symptoms of the patients were fever and abdominal pain, consistent with findings in most other studies (6,12). Vomiting, myalgia, joint pain, and chest pain were other common symptoms. Unlike many studies, no signs of skin rash were observed in our patients (20, 32). A less common clinical finding was febrile seizures observed in P3 (with genotype p.V726A/p. E148Q). Studies have reported that the frequency of febrile seizures grows in patients with FMF compared to the general population (33, 34). This intriguing finding, which may be due to high fever during FMF attacks in individuals predisposed to febrile seizures or could be a neurologic complication of FMF, opens up new avenues for research and potential implications for patient care.
Conclusion
These findings helped to identify mutational hotspots in the population, discover novel or rare mutations, and design a local molecular diagnostic strategy. Such research ensures faster and easier identification of patients and carriers, expediting treatment and preventing long-term complications. In this way, our study further confirmed the pathogenicity of the rare p.S339F mutation. In addition, in line with previous research from different populations, this study suggested the high frequency of potentially pathogenic mutation of p.R202Q. Besides, its association with the full spectrum of FMF features underscores the importance of including this mutation in the routine molecular screening of patients. Further studies with a larger sample size are needed to discover other MEFV gene mutations, confirm their clinical significance, and detect hotspot regions in different populations.
Declarations
Acknowledgements
This article is the result of Ms. Zohreh Keykhodaei's thesis, which she completed to earn a master's degree in Immunology from AJUMS, with support from the Cellular and Molecular Research Center, Ahvaz Jundishapur University of Medical Sciences (CMRC-9902). We appreciate the cooperation and trust of the patients and their families, as well as that of Ms. Atefeh Heydari.
Ethical Considerations
Under the Ethics Committee of Ahvaz Jundishapur University of Medical Sciences, the study was approved (Approval ID: IRAJUMS.MEDICINE.REC.1399.003). All participants signed an informed consent form following detailed explanations of the study's objectives and procedures.
Authors' Contributions
Project administration, P.G.; design of experiments, A.G.; supervised patients' clinical studies, M.F., A.K., and A.S.; planning and conducting experiments, P.G., Z.K.; writing—original draft preparation, Z.K.; editing, P.G.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Fund or Financial Support
Ahvaz Jundishapur University Medical Sciences funded this study (CMRC-9902).
Using Artificial Intelligence Tools (AI Tools)
Received: 2024/11/29 | Accepted: 2025/03/1 | Published: 2025/03/13
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy