BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://journal.zums.ac.ir/article-1-7270-en.html



2- Dept. of Pediatrics, School of Medicine, Ayatollah Mousavi Hospital, Zanjan University of Medical Sciences, Zanjan, Iran
✅ This study reports clinical laboratory, radiological, and magnetic resonance imaging (MRI) findings as well as whole-exome sequencing (WES) of a female patient aged 19 months and 7 days with encephalopathy. The limitation of this study such as lack of access to the patients or biological samples of other similar patients hindered further evaluation. Hence, comprehensive research must be conducted to reveal the underlying etiology.
Encephalopathy refers to a clinical condition characterized by ischemic stress-induced cerebral dysfunction which can result in multiple clinical symptoms. As a generalized disorder of brain function, encephalopathy can be acute or chronic and progressive or static. Scientifically, encephalopathy refers to diffused brain diseases capable of altering the brain structure or function. Several potential etiologies have been attributed to this disorder, including infection, metabolic disorders, brain tumors, increased intracranial pressure, malnutrition, and reduced oxygen supply of the brain (1). Previous studies revealed that encephalopathy has affected over 250,000 people in the United States. Inflammation of the brain parenchyma is the primary cause of encephalopathy, manifesting in various symptoms such as fever, headache, clouded consciousness, seizures, personality changes, focal neurological deficits, and even coma. Additionally, encephalopathy can occur alongside meningitis and inflammation of the membranes of the brain and spinal cord (2).
Acute encephalopathy (AE) is a neurological dysfunction that may cause consciousness alterations, neurological damage, or even death in young children (3).
Various etiologic causes are associated with AE, including infectious agents, toxic substances (such as carbon monoxide, drugs, lead, etc.), metabolic disorders (such as hepatic, uremic, diabetic ketoacidosis, etc.), ischemic events, hypertensive episodes, autoimmune disorders, inherited metabolic errors, and other unknown causes. AE is frequently observed in pediatric patients and has been linked to virus infections such as influenza (4-6), rotavirus, mumps virus, human herpes virus-6 (HHV-6), and the Picornavirus family. However, other potential etiologies should not be overlooked in pediatric patients (4).
Inborn metabolic errors are often associated with AE and can result in lower consciousness levels with severity ranging from minor changes in behavior to severe unconsciousness showing little or no response to stimuli. Identification of the exact cause of AE is challenging in the early stages. However, it is crucial to quickly diagnose the underlying cause of AE to ensure appropriate and effective management (7).
This study thus aims to evaluate a patient with AE who was admitted to Ayat Allah Mousavi hospital in Khodabandeh city, Zanjan province, Iran. The present study was conducted due to the high prevalence of AE with similar clinical symptoms in a specific area of Khodabandeh city. The main objective was to investigate the clinical and laboratory findings of the patient to identify the etiology of AE.
Patient information, including age, sex, clinical signs and symptoms, the disease course, the laboratory test results and radiology images, and also whole exome sequences, were extracted and evaluated. Blood samples (2 mL) were obtained from both patient and her familial guardians. Standard salting out protocol was utilized for the isolation of DNA from peripheral blood. The Nanodrop (Epoch, Biotek, USA) and 1.5% agarose gel were employed to determine the quality and quantity of the extracted DNA.
Sequence variants were annotated utilizing databases containing population and literature data, specifically 1000 Genomes (http://www.1000genomes.org/), GnomAD (http://gnomad.broadinstitute.org/), Online Mendelian Inheritance in Man (OMIM) (http://omim.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php). The variants were interpreted according to the guidelines provided by the American College of Medical Genetics (ACMG).
This double-blind randomized clinical trial included 64
Patient Demography and Symptoms
The female patient of this study aged 19 months and 7 days and was admitted to a local center due to cough, runny nose, and vomiting for two days. Following her last vomiting episode, she experienced a seizure, which w::as char::acterized by cyanosis, decreased level of consciousness, and hypotonicity. She was subsequently transferred to the Pediatric Intensive Care Unit (PICU) after undergoing Cardiopulmonary resuscitation (CPR) and intubation. During her hospitalization, she experienced repeated seizures in the form of limb spasticity, dizziness, blinking, and syrup of mucous muscles.
Review of her medical history revealed that she was the second child of the family and was born at term gestation with a birth weight of 2100 grams. There was no history of head trauma, and her parents were not relatives. Additionally, there was no significant past medical history or previous hospitalization. She had not taken any medications except for those administered during resuscitation (such as epinephrine and serum). Moreover, she was febrile during the initial examinations. She was intubated, had occasional spontaneous breathing, exhibited mydriasis of pupils (but was responsive to light), and her growth indexes were within the normal range.
Laboratory, Radiological, and MRI findings
The laboratory evaluations indicated mild anemia; her CSF, ESR, CRP, CXR, and echocardiography were within the normal range. Brain CT investigation and MRI analysis of the brain indicated brain edema and hypersignality, respectively (Figure 1 - 2). Furthermore, EEG showed diffused low-voltage slow waves due to encephalopathy.
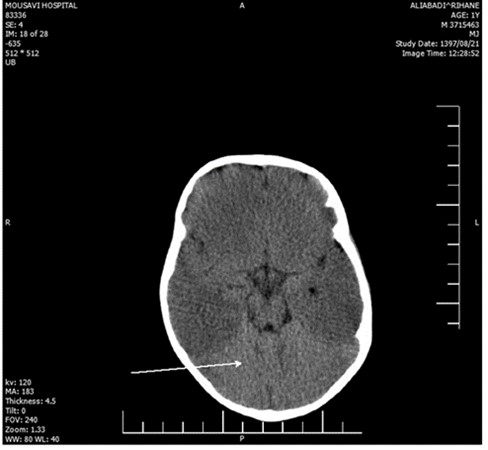
Figure 1. The CT of brain in current patient
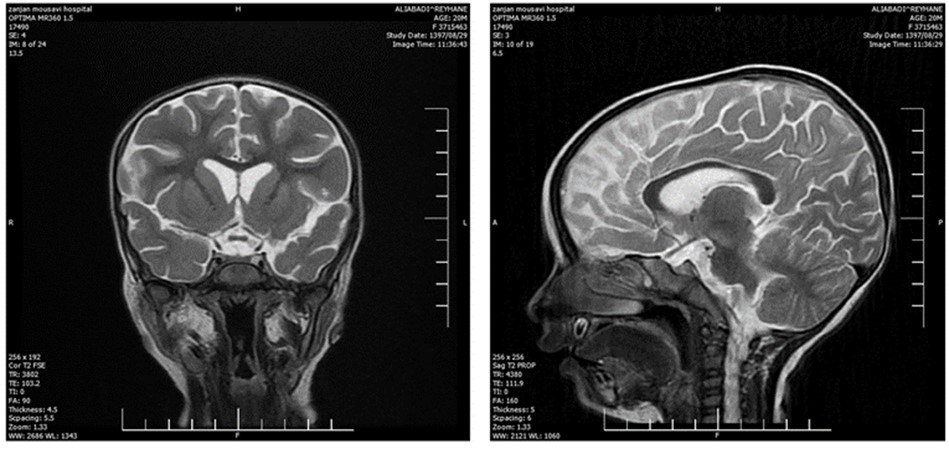
Figure 2. The MRI analysis of the brain
Whole- Exome Sequencing
Whole exome sequencing was performed on genomic DNA The c. 352G>A heterozygous variant in the PPP2RIA gene (exon four of chromosome 19) was diagnosed. This variant has not been previously reported as a pathogenic variant and was classified as a variant of uncertain significance (VUS) based on American College of Medical Genetics (ACMG) standards and guidelines. The Human Gene Mutation Database (HGMD) and ClinVar dataset were explored to assess phenotype relation and variant novelty through a review of resources. The BLOSUM62 score metric (1.0) indicated a moderate effect. Meta-SNP was utilized through various predictors (PANTHER, PhD-SNP, SIFT, and SNAP) to determine the effect of mutation on the normal protein. The overall prediction by Meta-SNP confirmed the reported mutation as a disease-causing variant, thereby, validating the results.
Patient follow-up
During hospitalization, her general condition improved with controlled seizures. She was removed from the ventilator (still hypotonia and decreased eye contact) and discharged after 21 days of hospitalization. Our follow-up at the age of two years and three months revealed that the patient had no capability to walk, grasp objects by hand, or speak (except for two words: mama and papa). However, she can sit down for a short time with help.
Discussion
The present study reports a case of encephalopathy in a pediatric patient. The primary objective of this study was to investigate the underlying etiology of encephalopathy, which can have multiple causes, including genetic origins. Several studies have also explored the unknown etiology of encephalopathy. For instance, Kwong and colleagues characterized 26 patients with infantile epileptic encephalopathy (IEE) of unknown etiology, with no specific epileptic syndromic phenotypes. Further genetic analysis revealed ten-point mutations, including ARX (1), CDKL5 (3), KCNQ2 (2), PCDH19 (1), SCN1A (1), STXBP1 (2), as well as one microdeletion involving both SCN1A and SCN2A. The high percentage of mutations (42%) suggests an underlying genetic etiology for IEE, and the authors proposed genetic evaluation for such patients (8).
In another study, Sartori et al. reported that the involvement of the pathogenic function of X-linked Cyclin-Depended Kinase-Like 5 and Aristaless-Related Homeobox genes of epileptic in refractory epilepsy with mental retardation (9). They also reported history of some casts of early-onset epileptic encephalopathy with lack of etiological clarification. Moreover, fumarase deficiency was reported as the cause of metabolic encephalopathy in a 5-month-old boy (10).
The PPP2R1A gene encodes the Aα regulatory subunit of protein phosphatase 2A (PP2A), which plays a crucial role in the regulation of various cellular processes in the brain. PPP2R1A mutations are associated with various neurological disorders, including childhood brain diseases such as epilepsy and intellectual disability according to Zhang et al (11). A De Novo variant in the PPP2R1 was reported as an infant induction of neurodevelopmental abnormalities. To summarize, they identified a new variant in the PPP2R1A gene with possible pathogenicity. Despite its rare incidence, this PPP2R1A-caused condition should be considered when dealing with patients with unexplainable neurodevelopmental abnormalities. Currently, adopting a variant-driven approach for unidentified disorders could be an effective method for determining the cause of potential genetic disorders.
Furthermore, Lenaerts et al. (12) reported the association of a wide range of phenotypes in PPP2R1A-related neurodevelopmental disorders with the extent of biochemical dysfunction. They documented 30 cases of PPP2R1A-related neurodevelopmental disorders, in which 16 different variants were identified. Out of them, 21 cases had variants that have not been previously reported. The severity of developmental delay varied from mild learning problems to severe intellectual disabilities with or without epilepsy. Language delay, hypotonia, and hypermobile joints are among the common symptoms. Macrocephaly was observed only in individuals without B55α subunit-binding deficit. Additionally, these patients had less severe intellectual disability and no seizures.
Taken together, these studies suggest that mutations in PPP2R1A can disrupt PP2A activity and lead to abnormal neuronal development, which may contribute to the development of childhood brain diseases such as epilepsy and intellectual disability. Further research is required to better understand the underlying mechanisms and develop targeted therapies for these disorders.
Further studies are recommended to address the identification of unknown etiology of encephalopathy. A previous study reported the frequency of antineuronal antibodies in patients with unknown etiology, which is associated with autoimmune encephalopathy. Additionally, developmental and epileptic encephalopathies (DEEs) have been attributed to genetic causes through novel DEE-associated genes, which support the diagnosis of denovo pathogenic mutations (13).
The variant of c. 352G>A in the PPP2RIA gene (exon 4 of chromosome 19) was detected in this study which has not been previously published as a pathogenic variant. Therefore, it can be considered a benign variant. Based on ACMG standards and guidelines, this viariant can be classified as a variant of uncertain significance (VUS).
Limitation
This case is one of the many patients with encephalopathy living in Khodabandeh city. It is evident that some patients have been referred to other hospitals in Zanjan areas, and even some were hospitalized in neighboring provinces. There is a possibility of death in some patients before the confirmed diagnosis of encephalopathy due to the severity of the initial symptoms and lack of access to imaging and other diagnostic procedures. In addition, this research, only addressed children with encephalopathy which may be similar to adult cases. Unfortunately, the medical files of some cases were lost due to the unavailability of their contact information. The parents of two patients that were not hospitalized in our hospital were not willing to participate in the study and did not cooperate. Therefore, we strongly recommend conducting a comprehensive study on this disorder in that area.
Conclusion
This study evaluated the clinical, laboratory, radiological, and genetic findings of a pediatric patient with encephalopathy. Although the patient had some common symptoms and laboratory findings associated with encephalopathy, a genetic variant in the PPP2RIA gene was recognized as the underlying cause. This finding highlights the importance of genetic testing in patients with encephalopathy, particularly in cases with unknown etiology. The timely identification and management of the disease can enhance the prognosis. Utilizing whole-exome sequencing may offer a reliable diagnosis and identify the classification. However, further research is required on multiple centers and a larger sample size to investigate the therapeutic approach for this condition.
Acknowledgements
This work was supported by Ethical No: ZUMS.REC.1401.214.
Authors' contributions
Hassan Bakhtiary conceptualized and designed the report, wrote the manuscript, and contributed to all stages of this case report. Narges Heidari was responsible for data collection, contributed to the drafting of the manuscript. All of authors read and approved the final manuscript. Hassan Bakhtiary is the guarantor of this study.
Funding
This study was performed without funding.
Conflicts of Interest
The authors declare that there is no conflict of interests.
Received: 2023/08/12 | Accepted: 2023/10/9 | Published: 2024/01/29
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy