BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
URL: http://journal.zums.ac.ir/article-1-6103-en.html



2- Social Welfare Organization of South Khorasan Province, Birjand, Iran
3- Cardiovascular Diseases Research Center, Birjand University of Medical Sciences, Birjand, Iran ,
✅ In conclusion, our study indicated a mutation in the PEX1 in the affected family. This mutation is a missense variant at codon 843 in ZS patients. It has an autosomal recessive inheritance pattern. This mutation may be widespread among Iranian population with ZS and can be used for a more desirable personalized medicine.
CPeroxisome is a ubiquitous cell organelle in human, which is essential for oxidation of branched-chain and very-long-chain fatty acids (VLCFAs), as well as the metabolism of plasminogens, amino acids, and ether lipids (1, 2). Peroxisome biogenesis disorders (PBDs) are a spectrum of diseases with peroxisomal defect and diverse peroxisomal metabolic dysfunctions due to mutations in the PEX genes (2,3). Reports demonstrated that the majority of Zellweger syndrome (ZS) cases have different mutations in the PEX1, PEX6, PEX10, PEX12 and PEX26 genes (4). ZS, also recognized as cerebrohepatorenal disorder, is a rare genetic condition which is defined by the absence or reduction of functional peroxisomes in the cells which are responsible for VLCFA beta-oxidation (5). Zellweger spectrum disorder (ZSD) and rhizomelic chondrodysplasia punctata type I (RCP) are classified into two categories. In addition, ZSD is categorized into three classes: neonatal adreno leukodystrophy (NALD) (the most common phenotype), infantile Refsum disease (IRD) and ZS (6). Epidemiological evidence shows that the ZS prevalence in the United States is estimated to be 1:50,000 births. Patients with serious disorders die in their first year of existence (7). However, a number of patients with milder phenotypes may survive until the second decade (8). A wide range of symptoms such as enlarged liver, renal cysts, seizures, retinal dystrophy, and hearing loss may be manifested (9).
Essential proteins for peroxisome biogenesis and conservation are called peroxins (PEX proteins). The most recognized peroxins promote protein entry from cytosol synthesis location to the peroxisome matrix. PEX1 ATPase promotes regeneration of protein receptor PEX5 peroxisome complex, which is the most frequently involved peroxin in biogenesis diseases of mammalian peroxisome. Mutations in the PEX1 are the frequent cause of PBDs in mammalian. In the lethal cases, the peroxisomes are not developed or the matrix proteins ingest ineffectively.
Therefore, a precise understanding of the molecular aspects of rare inherited disorders, such as ZS, requires genomic sequencing as a novel and effective strategy (10, 11). Presently, the whole-exome sequencing (WES) technology and the survey of protein-coding regions of genes in a genome, provide feasibilities for ZS investigation (12, 13). Herein, we aimed to present the molecular diagnosis of ZS in an Iranian family in the South Khorasan Province, using WES to detect the accurate etiology of idiopathic ZS.
Informed Consent and Ethics Guide
All participants from each family signed the informed consent, and the entire procedure was performed according to the tenets of the Helsinki Declaration. The ethics code was approved by the Institutional Research Ethic Committee of Birjand University of Medical Sciences (Ir.bums.REC. 1396.350).
Clinical Evaluations
The participated family included father, mother and one female proband who were supported by the Social Welfare Organization of South Khorasan Province, Iran. Total siblings consisted of one affected brother (twin) and four unaffected ones. Peripheral blood samples were obtained from all family members.
Whole-exome Sequencing Analysis
Proband Genomic DNA (gDNA) was extracted via standard salting out method (14-16). The quantity and quality of extracted DNA were determined by Nanodrop (Epoch, Biotek, USA), and 1.5% agarose gel. In this setting, WES was performed on the DNA sample of the affected twin and the parents by a HiSeq2000 instrument (Illumina, San Diego, CA) following the manufacturer's instructions. The mapped reads were aligned to the human genome sequence (UCSC GRCh37/hg19), and homozygous genotypes were filtered based on dbSNP as shown in the UCSC Genome Browser, HapMap project, and 1000 Genomes Project variant database.
The effects of protein sequence substituted on the molecular effects of variants were studied. To determine the accuracy of deleterious variant prediction, some in silico software tools, including SIFT, PANTHER, PolyPhen and PROVEAN were employed. According to the outcomes of the cited procedure, the variant effect on protein function was predicted based on the protein sequence and structure. Wild-type and mutant PEX1 predictions of the three dimensional structure were performed by Iterative Threading Assembly Refinement (I-TASSER) server.
Also, in order to determine the conclusive connection between PEX1 (wild and mutant) and PEX6, as a component of the PIG family, molecular docking was achieved. HEX server calculated the interaction energy of docking (http:/www.loria.fr/~ritchied/hex/).
DNA Sanger sequencing analysis was performed to confirm the variant effect on the disease. A pair of primers were designed for polymerase chain reaction (PCR) amplification and sequence analysis (Table 1).
Table 1. The primers sequence which were used to confirm PEX1 mutation
| Product Size | Self-End | Self Any | GC% | Tm | Len | Sequence | |
| 367 | 3.00 | 4.00 | 46.43 | 65.41 | 28 | GCGGGTTTGTGGCTAGAGATTTTACAGT | Forward Primer |
| 4.00 | 6.00 | 50.00 | 65.54 | 28 | CTACTCTCTCGTGCAATTACCCCAGCTA | Reverse Primer |
Clinical Outcomes
Two eight-year-old (twin) affected individuals were enrolled in the study. They belonged to the Persian race, from the South Khorasan Province, southeast of Iran. The probands with ZS were born to a fifth gestation (Figure 1). Nervous abnormalities, such as intellectual disability, speech difficulty, and seizure appeared. The patients had normal weight, height and head circumference at the time of birth (Figure 1). According to the pedigree analysis, family history, and transmission pattern, the autosomal recessive inheritance was suggested for the current family.
Whole-exome Sequencing Findings
The proband, an eight-year-old female, had a PEX1 (NC 000007.14) missense mutation C.G2528A. It happens at the chromosome location of 7q21.2 which consists of 24 exons. This genomic mutation was confirmed by Sanger sequencing analysis of the affected proband female and her twin brother (Figure 1). No additional variations that could be a pathogenic candidate have been detected. Other population databases such as ClinVar dataset, Exome Sequencing Project (GO-ESP), Human Gene Mutation Database (HGMD), and 1000 Genomes project have reported this variant. In the primary level of variant filtering, we studied the missense, nonsense and frameshift indels. Further impacts of deleterious variants were kept on the stable secondary structure and protein folding. Based on the pedigree analysis, the homozygous variants were indicative of the autosomal recessive pattern of inheritance. Variants with a minor allele frequency (MAF)>1% were removed based on dbSNP links and more variant databases. The PEX1 gene, which is known to cause PBDs, was selected. A missense mutation was identified in exon 15 (22 out of 60 alleles) of the PEX1 gene. The c.2528GRA is in the second functional AAA (D2) domain in patients with PBD. Exon 15 sequencing of the PEX1 gene revealed homozygosity in the affected twin, and heterozygosity in the parents. DbSNP, 1000 Genomes Project, ExAC and Clinvar databases have identified the missense version. However, this mutation was absent in Iranian reports. The pathogenic related variants of PEX1 gene are presented in Table 2.

Figure 1. Identification of a missense PEX1 mutation. a) The proband indicated that specific characterize of Zellweger Syndrome in 8 years old including facial dysmorphology, speech difficulties and mental retardation; b) Consanguineous pedigree displayed affected members are II-4 and II-5 (twin). They are homozygous for the C.G2528A variant.
Table 2. The related Pathogenic variants of PEX1 gene
| Number | Variant ID | Variation (location) |
Variation type | Condition(s) | Clinical significance (last reviewed) |
Reference |
|---|---|---|---|---|---|---|
| 1 | 22557 | c.1906_2064del159 (p.Arg636_Leu688del) | Deletion | Zellweger syndrome | Pathogenic (Apr 14,1998) | (28) |
| 2 | 217431 | c.3750G>A (p.Trp1250Ter) | Single nucleotide | Deafness enamel hypoplasia nail defects | Pathogenic (Oct 1,2016) |
(29) |
| 3 | 488572 | c.3579delA (p.Asp1194Ilefs) | Deletion | Zellweger syndrome | Pathogenic (Nov 16,2017) |
(30) |
| 4 | 371698 | c.3574C>T (p.Gln1192Ter) |
Single nucleotide | Zellweger syndrome, Deafness enamel hypoplasia nail defects, Peroxisome biogenesis disorder 1B | Pathogenic (Feb 2,2017) |
(31) |
| 5 | 167443 | c.3505_3517delCAGTTGTTTTCAC (p.Gln1169Serfs) | Deletion | not provided | Pathogenic (Feb 11, 2014) |
(32) |
| 6 | 561079 | c.3205C>T (p.Gln1069Ter) | single nucleotide variant | Zellweger syndrome | Pathogenic (Sep 1,2017) |
(33) |
| 7 | 224325 | c.2966T>C (p.Ile989Thr) | single nucleotide variant | Zellweger syndrome, Deafness enamel hypoplasia nail defects | Pathogenic (Oct 1,2015) |
(34) |
| 8 | 188729 | c.2926+1G>A | single nucleotide variant | Zellweger syndrome | Pathogenic (May 25, 2016) | (35) |
| 9 | 371696 | c.2922delA (p.Leu974Phefs) | Deletion | Zellweger syndrome, Peroxisome biogenesis disorder 1B | Pathogenic (Jun 16, 2016) |
(36) |
| 10 | 7516 | c.2528G>A (p.Gly843Asp) |
single nucleotide variant | Zellweger syndrome, Deafness enamel hypoplasia nail defects, Peroxisome biogenesis disorders, Leber congenital amaurosis | Pathogenic (Feb 6, 2015), Pathogenic (May31,2018) |
(37, 38) |
Discussion
We have detected a missense variation in PEX1, in an Iranian ZS patient. The proband had the common characteristics of ZS including facial dysmorphology, difficulty in speech and intellectual disability. Since the clinical appearance was not trustworthy, the affected individual was first distinguished based on the symptoms. The pedigree analysis demonstrated an autosomal recessive pattern (Figure 1). Subsequently, based on the WES findings of the proband, a missense variant (C.G2528A) was detected, with an autosomal recessive pattern in the second functional AAA domain (D2) (Figure 1).
PEX1 (MIM#602136) is a protein-coding gene located on chr7q21.2 location, which contains 24 exons. It produces a transcript of 4.4 kb. PEX1 mutations are detected in almost 70% of ZS patients. Evidence suggests that PEX1 can play a critical role as a matrix protein transporter in the peroxisomal membrane. The protein structure generally consists of four segments: N-terminal and C-terminal regions, and D1 and D2 domains. To date, 14 human PEX genes have been identified which encode peroxin proteins. They have function in different phases of peroxisome biogenesis, such as peroxisomal matrix protein importation, membrane formation, and peroxisome proliferation (17, 18). Peroxisomal membrane proteins are active as signaling sites for the autophagy, which are caused by antiviral immunity and reactive oxygen species (19). Glycine (Gly), a non-charged amino acid, is substituted with Aspartate (Asp) which is negatively-charged, and this could induce crucial functional and structural alterations. Phylogenetic protein sequence conservation analyses of PEX1 reveals that Glycine 843 is a protected amino acid in the protein sequence of human PEX1 (Figure 2). However, the exact protein function is not clearly known.
In a patient with Leber congenital amaurosis (LCA), the mutant PEX1 (p.Gly843Asp) which has a transformation (Gly to Asp) in cDNA bp 2528 has been documented once before (20). Reuber et al. reported that mutations in PEX1 could be the most common cause of PBDs occurrenceThey indicated that PEX1 deficient cells have different deficiencies in the peroxisomal targeting signal and protein matrix transfer. Figure 3 displays the projected functional partners of PEX1. Moreover, their results showed that the G843D allele is a common mutation in the PBDs (21). The new mutation attenuates the protein function and is associated with mild peroxisomal condition of group 1 complementation (Figures 4 and 5).
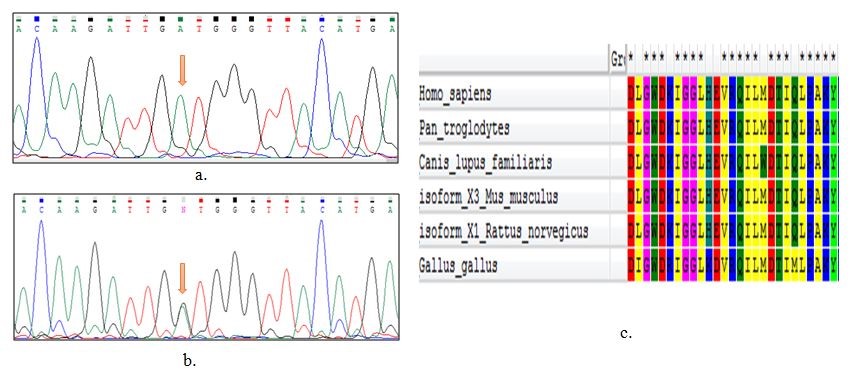
Figure 2. a) The Sanger sequencing analysis of homozygote affected. b) The Sanger sequencing analysis of heterozygote parent, an arrow shows the mutation; c) phylogenetic conservation studies of the PEX1 protein sequence and Glycine is conserved amino acids in the human PEX1 protein sequence
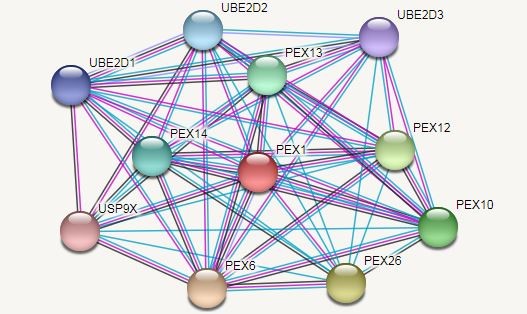
Figure 3. Predicted functional partners of PEX1
.jpg)
Figure 4. Prediction of PEX1 protein structure. To comprehend, the structural implications of mutations in PEX1, wild-type and mutant proteins were modeled using the online I-TASSER server.
.jpg)
Figure 5. Docking proteins to find the effect of mutation on PEX1
Meng-Meng Ge et al. reported four novel mutations (c.2416+1 G>T and c.2489delT) (c.1483+1 G>A and c.1727dupG) in the PEX1 gene using the WES technique (22). Ling Yu verified the homozygous mutations (c.818insCTTG) (p.Pro274Leufs*8) in the PEX6 gene by WES, which target gene panels of 'multiple congenital anomalies' and 'skeletal dysplasia’ in ZS patients. Evidence showed that the two novel mutations (p.Cys358* and p.Leu83Pro), have been known as pathogenic according to Genomics guideline. Phenotype-genotype associations were reported in the PBD-ZSD continuum patients (23). Barillari and coworkers recognized two specific variants in PEX1 by sequencing. One of them was the missense variant p.(Val92Leu);c.274 G>C which has been mentioned in the PBD cases before. The second one was p.(Ser714 Gln715dup);c.2140 2145dupa, which is a non-frameshift variant. It is not found in normal samples (24).
The researchers found that, in two Chinese newborns, all four mutations have critical control over the protein function. In addition, evidence indicates that the main causes of multiple organ dysfunction and neural disorder are β-oxidation deficiency, VLCFA accumulation, and docosahexaenoic acid (DHA) reduction (22). Bousfiha et al. detected a new homozygous PEX1 mutation (p. Leu1026Pro) in two deaf Moroccan siblings. Their reported symptoms included vision impairment, deep deafness and delay in development. They showed that this mutation can be because of ATP hydrolysis in the P-loop (25).
Recent developments in high‐throughput WES approaches and the next‐generation sequencing method have now revealed the genetic basis of disorders with unknown etiology. Furthermore, exome sequencing is progressively being used as an investigative device for precise genetic diseases diagnosis, especially in the context of substantial phenotypic and genetic heterogeneity disorders (26, 27). This result can be replicated as a result of consanguineous marriage in the future, which can be used for a more suitable customized treatment for ZS patients.
Conclusion
The results of the present study indicated a pathogenic heterozygous missense mutation in PEX1 at the codon 843 in ZS patients. This finding can be repeated in the future in the cited family. It may happen as a result of consanguineous marriage. This mutation may be used for ZS patients for a more desirable personalized medicine. Additionally, it is a missense variant (p. Gly843Asp) with an autosomal recessive pattern. It may be widespread among Iranian populations with intellectual disability. Taken together, the results of this study provided novel perceptions into the diagnostic power of WES, and revealed that it is possible to consider and treat the recognized mutations, individually.
Funding and support
This research resulted from an independent research without receiving any financial support.
Conflicts of Interest
Authors declared no conflict of interests.
Received: 2020/07/5 | Accepted: 2020/09/15 | Published: 2020/12/30
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |
Copyright Policy